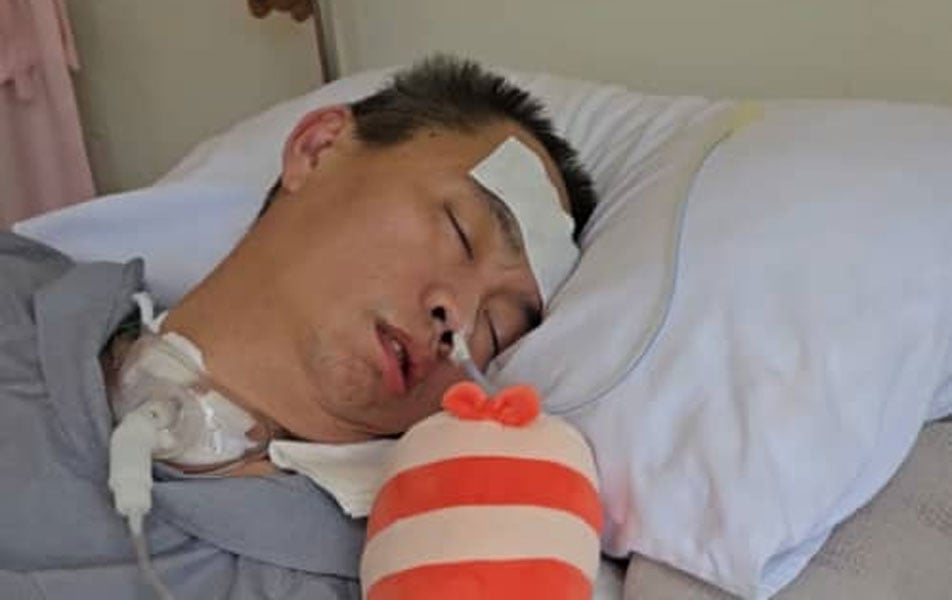
余福与李佩玲盼求大众捐助尚缺的医费,让余捷申能在接下来的两年继续用药,右为蔡瑞豪。
8岁男童患上罕见基因病——脊髓性肌萎缩症,其身体肌肉会逐渐萎缩,等了6年终盼得有药物注射以延缓萎缩,父母求助所欠缺7万5198令吉供两年医疗费用。
来自吉隆坡文良港的余捷申自胎儿期至出生一切正常,直到18个月大时,妈妈发现他没有像其他孩子一般正常爬行及走路,辗转从儿科转介到脑神经科以及基因科后,才于9个月后,即小捷申2岁时,被诊出患上脊髓性肌萎缩症(SMA TYPE 3)。

男童父母东借西筹之下让小捷申注射1年的药物,随后能站起来走路、游泳等,且体能明显增强,让原本无计可施的父母重燃希望。然而,每年药物费用为6万2599令吉,他们的经济能力每年仅能负担2万5000令吉,遂向大山脚瑶池金母慈善基金(ONE HOPE CHARITY)求助。
该基金会主席拿督蔡瑞豪周三说,小捷申行动缓慢,每次行走一会儿就会累,而且走路不稳容易跌倒、上下楼梯都是手脚并用,上学时都是坐着电动轮椅出入学校,也会游泳,通过水的浮力来强化其下肢。
他指出,自去年开始注射药物后,小捷申的体能大幅改善,已经可以站立,走路较稳健不跌倒,连游泳也可以从15米两圈提升至10圈,进展非常良好。
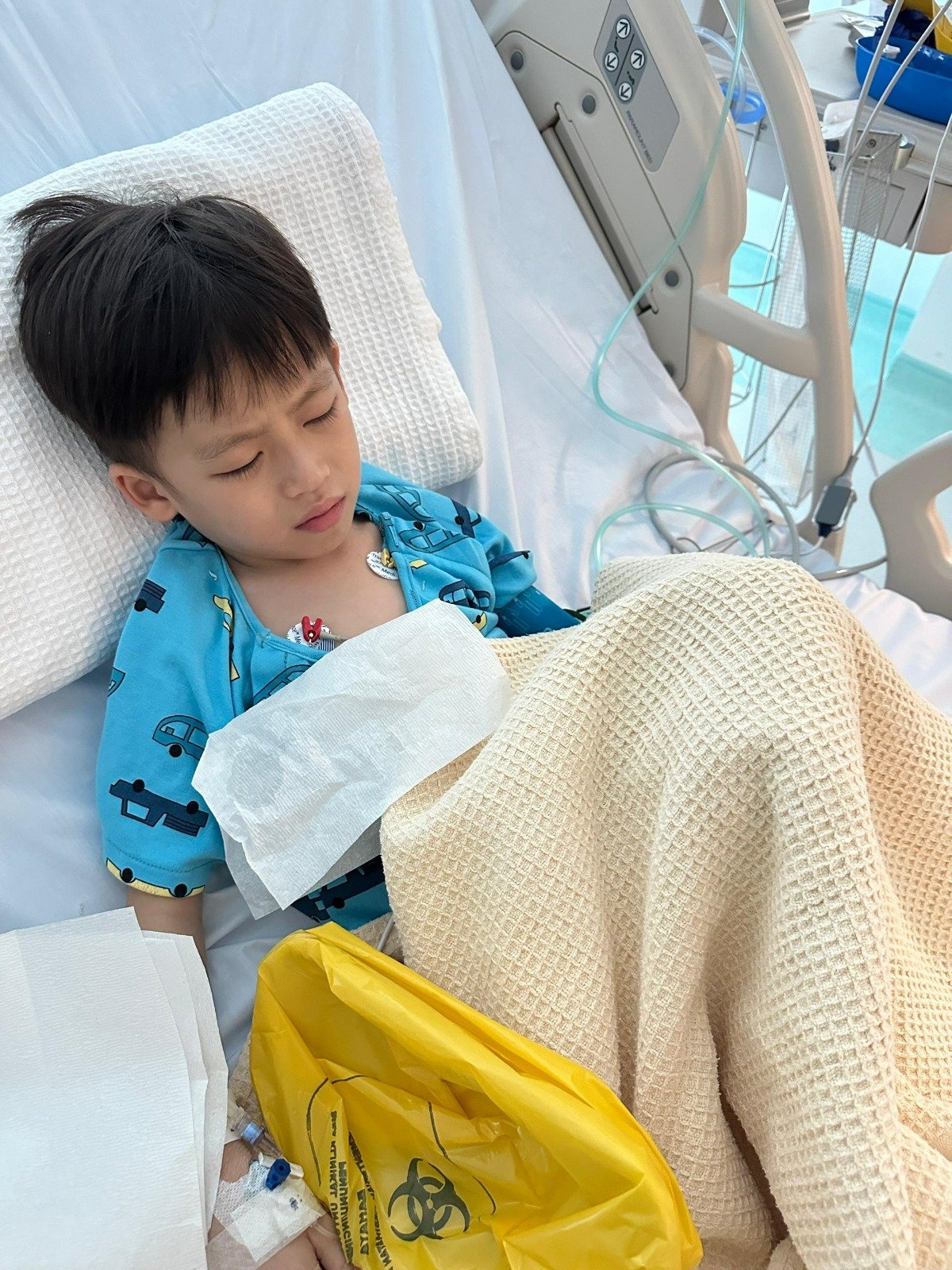
“除了罕见基因病,小捷申在幼儿时期全身痛兼发烧不退,检验后证实他患上自身免疫性疾病(Autoimmune disease),这个疾病会攻击小捷申的关节、心脏、皮肤等,导致其关节痛、出红疹及脱皮,医生告知这需要服用免疫药物,直到他21岁为止。”
男童父亲余福(39岁)及母亲李佩玲(39岁)皆是房产经纪,两人共育有4名孩子,年龄介于6岁至13岁之间,该罕见疾病是四分之一的遗传机率,在4名孩子中,一人正常,两人是带原者,只有排行第三的小捷申病发需要用药。
大山脚瑶池金母慈善基金官网:www.onehopecharity.org 及热线016-4192 192、019-2322 192、04-539 9212。

“我以为孩子走路不稳,可以透过药物或者手术医治,当检验后被告知这是万分之一机率的基因问题,无药可医让我们深感绝望。时隔多年后终盼到有药可治,恳求社会大众的帮助,让孩子拥有用药延缓肌萎缩的机会。”
李佩玲坦言,当初医生告知孩子患上不治之症,让他们深感晴天霹雳,当时因无药可医,孩子只能通过物理治疗、游泳等延缓萎缩,但治标不治本。
她说,当时全球有关反义寡核苷酸疗法(Spinraza)药物是天价药,即每年需要75万美金(折合约马币300多万令吉)的药物费,他们对此一筹莫展,根本负担不起而被迫放弃治疗。
“2023年这款药物正式被引入大马,连价格也降低后,我们重燃治疗的希望,也东借西筹尝试让孩子第1年施打6剂药物,如今孩子进展良好,可以继续用药。”
小词典:
脊髓性肌萎缩症(Spinal Muscular Atrophy, SMA)是一种由SMN1基因缺陷引起的神经肌肉疾病,是一种遗传性神经肌肉疾病,主要影响脊髓中的运动神经元,因控制肌肉运动的信号传递中断导致肌肉无力及萎缩,通常从四肢和躯干的近端肌肉开始,病情可能影响到吞咽、呼吸等功能。
根据发病年龄和严重程度,SMA分为4类型:SMA 1型最严重,主要发病期介于1至6个月婴孩期,出生后肌肉无力、无法坐起、呼吸困难、甚至影响吞咽问题,预后较差。
SMA 2型发病期介于6个月大至18个月大,幼儿可以独坐但无法站立,疾病进展缓慢,常见呼吸及骨骼畸形问题。
SMA 3型则是发病期超过18个月,病人可站立及行走,但随着年龄增长而失去行走能力。SMA 4型发病期则是30岁以后,是最轻微的症状,主要是表现在轻度的肌无力和肌肉萎。
尽管它曾被认为是一种无法治愈的疾病,但近年来的基因疗法和创新药物已显著改善患者的预后和生活质量。
找工作, 就找这里!


- Social Media Marketing Executive
- Advertising & Marketing
- Kuala Lumpur
-
 MYR 6K /Month
MYR 6K /Month

- PHP Software Developer
- Information Technology
- Wilayah Persekutuan
-
MYR 6K /Month